Access valuable RNA-seq insights in mere hours
Want to take the stress out of data analysis and fast-track your path to gene expression insights? With our new RNA-seq Analysis Portal, go from FASTQ files to pathway analysis insights in hours instead of days, integrating your data with qPCR and dPCR verification.
Easy. Fast. Online. Our portal supports the analyses for 18 different species using proven published algorithms. The RNA-seq Analysis Portal is compatible with QIAseq and other major RNA library kits. Perform your analyses anytime, anywhere.
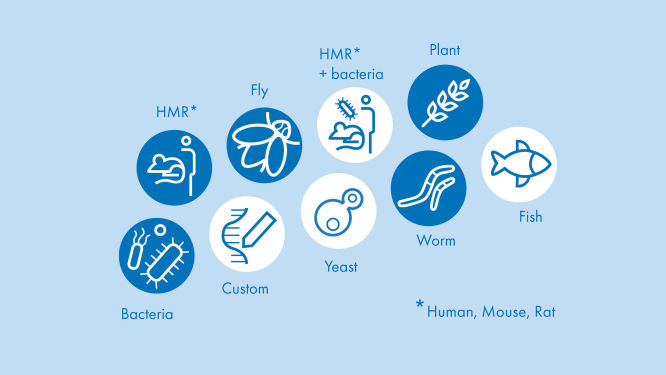

Support for the most commonly used RNA library kits
The RNA-seq Analysis Portal is compatible with the most commonly used RNA library preparation Kits.
Illumina TruSeq Stranded Total RNA Library Prep (Human/Rat, Gold, Globin) AND Stranded Total RNA Prep with Ribo-Zero Plus
New England Biolabs NEBNext Ultra II Directional RNA Library Prep Kit for Illumina Roche Sequencing solutions
QIAGEN QIAseq miRNA Library Kit, QIAseq UPX 3’ Transcriptome Panel, QIAseq UPXome RNA Library Kit, QIAseq Stranded RNA Kits, QIAseq FastSelect Kits
KAPA / Roche RNA HyperPrep Kit
Takara Bio SMARTer Stranded Total RNA Sample Prep Kit – HI Mammalian AND SMARTer Stranded Total RNA Sample Prep Kit – Low Input Mammalian
Thermo Fisher Scientific Collibri Stranded RNA Library Prep Kit for Illumina Systems
Testimonial from our global partner

"The RNA-seq Analysis Portal displays all the necessary information on a single screen in an easy-to-understand manner, making it easy to analyze and draw the plots and create figures which can be used for publication."
Dr. Noriaki Sasai, Developmental Biomedical Science, Division of Biological Science, Nara Institute of Science and Technology, Ikoma, Japan
Learn how RNA-seq data analysis can help you with your research


Take complete control of your RNA-seq data analysis

FAQ
Which RNA library preparation kits can I analyze with the RNA-seq Analysis Portal?
What species can I analyze with the RNA-seq Analysis Portal?
Can I analyze single-cell data with the RNA-seq Analysis Portal?
How is RNA-seq data analyzed?
How is primary RNA sequencing data analysis performed?
How is secondary RNA sequencing data analysis performed?
How is tertiary RNA sequencing data interpretation performed?
How should I normalize my raw RNA sequencing data?
How should I set up my RNA sequencing-based differential gene expression analysis?
What are fold change values and how are fold change values calculated?
What are p-values and how are p-values calculated?
Introducing RNA Sequencing-Based Differential Gene Expression Analysis with the RNA-Seq Analysis Portal
The RNA-seq Analysis Portal is an online RNA sequencing analysis software application designed for biological researchers with little or no bioinformatics experience or expertise. Its RNA sequencing analysis pipeline is fixed but optimized so you don’t have to worry about the primary RNA sequencing analysis details. The RNA-seq Analysis Portal also completes the secondary differential gene expression analysis calculations of fold-change values and p-values. The RNA-seq Analysis Portal also provides RNA-seq data visualizations such as a volcano plot and a heatmap, both commonly used to report differential gene expression analysis results in peer-reviewed publications. All you need to focus on is setting up the sample comparisons that you need to make.
With the RNA-seq Analysis Portal, you can analyze your raw RNA next generation sequencing data for your gene expression-based biomarker discovery – or any other project – with ease and confidence. It supports seven of the most frequently used RNA library preparation kits and 18 of the most commonly studied species model systems. Move immediately from your instrument’s FASTQ files to raw gene expression values. Fixed but optimized pipelines are available specific for the RNA library preparation kit that you used. Transcriptomes for each of the supported species are also available to automatically map reads to genes.
The RNA-seq Analysis Portal automatically takes the raw gene expression values and calculates fold change values and p-values using a published and well-established normalization method to correct for any experimental variation from nucleic acid isolation through the sequencing run. You are then free to use the fold change value and p-value filters as well as the automatically updated volcano plot and heatmap visualizations to find a subset of statistically significant differentially expressed genes. The application’s “What’s Next” feature allows you take that preliminary biomarker panel and further validate it using an alternative method such as real-time or digital RT-PCR experiments and a larger number of samples.
The volcano plot automatically generated by the RNA-seq Analysis Portal graphs for each gene the log2 of its fold change value on the x-axis versus the log10 of its p-value on the y-axis. A log2 fold change value on the x-axis of zero means no change in gene expression. Up-regulated genes return positive, while down-regulated genes return negative log2 fold change values on the x-axis. Larger numbers log10 p-value y-axis values indicate greater statistical significance. The most statistically significant differentially expressed genes of interest lie in the upper left and right sections of the volcano plot beyond researcher defined thresholds.
The heatmap automatically generated by the RNA-seq Analysis Portal is a non-supervised hierarchical clustering of selected normalized gene expression values across all samples. Genes are clustered along one axis, while samples or groups are clustered along the other axis. More intense color coding indicates lower or higher normalized expression values for each gene across the samples or groups compared. Samples should cluster into their defined experimental groups. Genes that have similar patterns of gene expression changes across the samples or groups then cluster together. Results may be used to build the classifiers that determine to which groups newly analyzed samples belong based on the expression of the genes selected and defined by the preliminary experimental analysis.
Setting thresholds for fold change value and p-value filters is crucial for interpreting any differential gene expression analysis. Transcriptome analyses can return thousands of differentially expressed genes. This number of targets is too many to be analyzed conveniently by real-time or digital RT-PCR. They may also be too many to analyze affordably with a focused or targeted RNA sequencing panel. Increasing fold change value and p-value threshold stringency decreases target numbers to only the most statistically significant and differentially expressed genes and those more likely to distinguish biological phenotypes. The RNA-seq Analysis Portal allows to easily and conveniently adjust fold change and p-value thresholds and immediately see the reduced number of gene targets in its table, volcano plot, and heatmap.
By combining all these features into one application, the RNA-seq Analysis Portal allows biological researchers like yourself to focus on the final differential gene expression analysis results. Simply ensure you have included appropriate control samples and multiple replicates for each of your biological conditions, including the controls. Then, move immediately to defining the sample or group comparisons you need to make.
Make the RNA-seq Analysis Portal an integral part of your RNA sequencing based differential gene expression analysis project today.